

大皖新闻讯 铜,是生活里随处可见的金属材料,它同样也是人体不可或缺的微量元素。但对一部分人而言,这份“必需”却成了危及生命的负担。
安徽的王先生兄弟俩,就不幸遇上了这样一种罕见遗传病——肝豆状核变性,身体无法自主代谢铜元素,脏器被铜离子慢慢堵塞侵蚀,兄弟二人先后走到了肝衰竭的生死边缘。
幸运的是,在中国科大附一院(安徽省立医院)刘连新教授带领下,该院肝移植科主任张树庚教授团队全力救治,三年前为弟弟实施肝移植手术挽回生命,近日又顺利为哥哥完成移植手术,接连帮助兄弟二人挣脱死神阴影,为这个深陷病痛的家庭守住了生机。
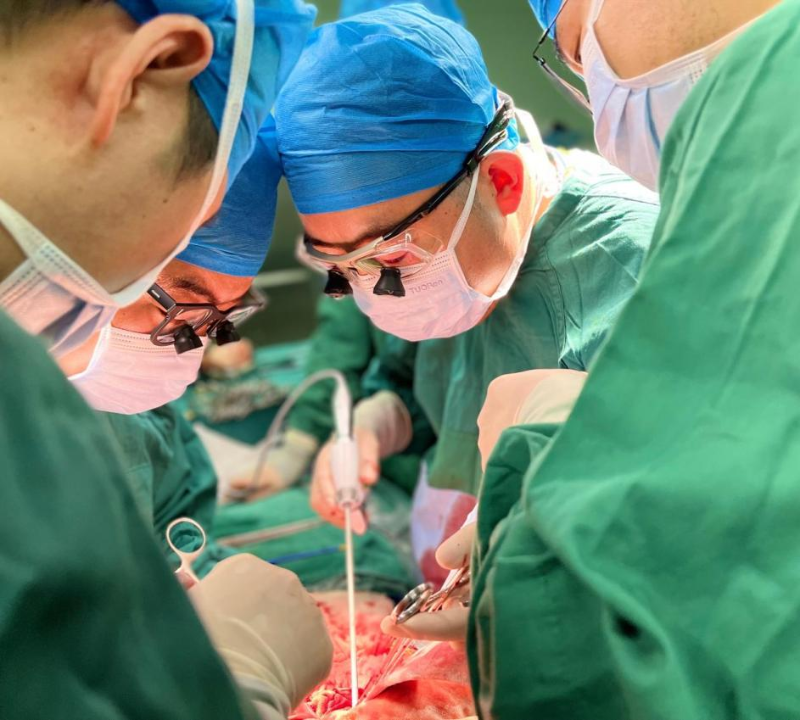
同病相怜:兄弟二人先后陷入肝衰竭绝境
据了解,王先生兄弟俩所患的肝豆状核变性,是一种罕见的常染色体隐性遗传的铜代谢障碍性疾病,患者的身体无法正常排出多余的铜,导致铜离子在肝脏、大脑、肾脏等器官中不断沉积,像水垢一样“腐蚀”脏器功能。
随着病情恶化,兄弟俩先后发展到肝功能失代偿期,肝脏正常代谢、解毒、合成功能严重受损,出现了黄疸、腹水、肝功能衰竭等危重症状。常规药物已经无法逆转病情,肝移植成了唯一的生路。
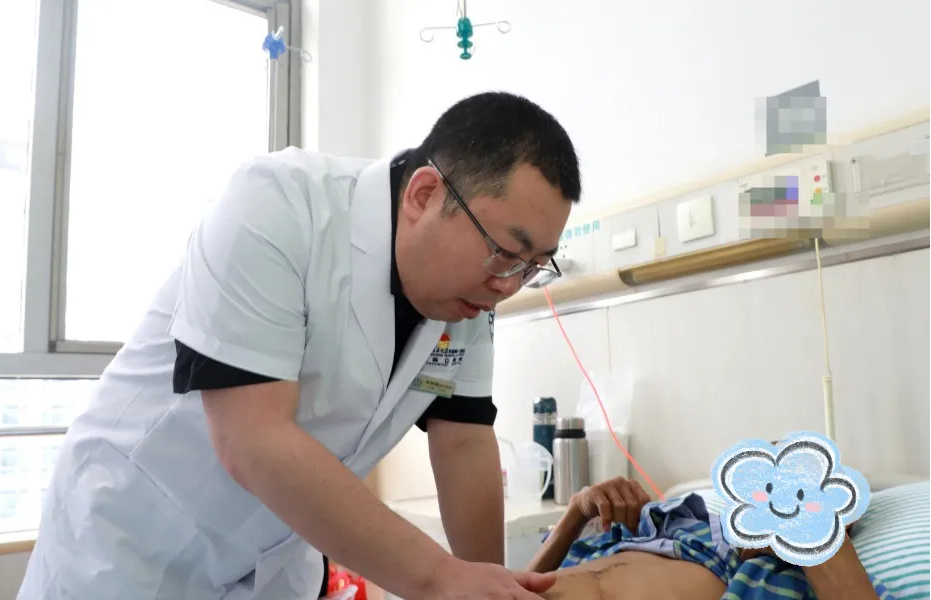
三年前,王先生的弟弟在中国科大附一院(安徽省立医院)成功接受了肝移植。看着弟弟顺利恢复,一天天精神焕发,重新回到了工作和生活中,王先生备受鼓舞。在弟弟的支持下,不久前,他也在该院肝移植团队的精心救治下,顺利完成手术。目前他恢复良好,各项指标趋于正常,已顺利出院。
追根溯源:基因缺陷让身体排铜系统“罢工”
好好的身体排铜系统,为何会出现“故障”呢?根源其实藏在一种基因缺陷中。
据介绍,肝豆状核变性的致病原因,是人体13号染色体上的ATP7B基因发生突变。这个基因负责编码一种“铜转运蛋白”,相当于身体里的“铜转运管家”。一旦管家罢工,多余的铜排不出去,就会在全身到处沉积。
铜元素长期在肝脏、大脑、肾脏等器官中异常蓄积,会持续侵蚀器官组织,逐步造成多系统、不可逆损伤:肝脏逐渐硬化、肝功能衰竭;脑区损伤引发肢体震颤、言语不清、精神异常;眼角膜出现特征性的“K-F环”;肾脏功能也会受损。
然而,由于大众对这种罕见病知晓度较低,加上发病症状繁杂多样,有些患者甚至直到肝衰竭阶段才被明确病因,从而错过了早期干预的时机。
破除误区:这种罕见病并非不治之症
很多人谈及罕见病便心生恐惧,认为其无药可医。但其实,肝豆状核变性并非不治之症,关键取决于发现和干预的早晚。
对于脏器尚未发生不可逆损伤的早期患者,通过规范的驱铜药物治疗和严格的饮食管控,能够有效清除体内多余铜元素,阻断病情进展,绝大多数患者可以长期稳定生活。
但一旦发展到肝功能严重失代偿阶段,出现急性肝衰竭、严重肝硬化、顽固性腹水等症状,常规驱铜药物已无法逆转肝脏器质性病变,此时及时开展肝移植手术,才能从根源上重建人体正常的铜代谢功能,解决排铜障碍,快速逆转病情,挽救晚期重症患者生命,提升远期生存率与生活质量。若错过移植的最佳时期,患者很可能因肝衰竭或多器官衰竭而失去生命。
科学防控:早筛、规范治疗与长期管理缺一不可
如何才能早期发现这种罕见病呢?专家提醒,对于5-35岁出现不明原因肝功能异常、肢体震颤、性格突变、学习能力骤降的人群,以及有家族遗传史的家庭,需及时开展铜代谢筛查、基因检测,实现早发现、早确诊。
一旦确诊,患者需在医生指导下长期规律服用驱铜药物,定期复查铜蓝蛋白、肝功能、神经系统指标,根据病情调整治疗方案,切勿擅自停药、减药。
同时,确诊患者还需严格管控饮食,禁食坚果、巧克力、动物内脏、贝类等高铜食物,日常选用低铜食材,减少铜元素摄入。
对于已出现肝功能失代偿、急性肝衰竭、难治性肝硬化的重症患者,需尽早评估肝移植指征,及时开展手术干预,这是救治晚期肝豆状核变性患者的核心方案,也是挽救重症患者的关键手段。
此外,患者需要长期随访,做好心理疏导,缓解疾病带来的心理压力。对于家族携带者,可以通过婚前、孕前基因筛查,提前规避遗传风险,阻断家族遗传链条。
大皖新闻记者 叶晓 张婉馨 通讯员 齐灿 方雯 汪志豪配资炒股平台官网
天创网提示:文章来自网络,不代表本站观点。


